J Am Soc Echocardiogr. 2004 Oct;17(10):1073-6.
Echocardiographic features of atrial myocarditis with giant cells: a case report.
Law KK, Binder J, Cooper LT, Specks U, Tazelaar HD, Seward JB.
Division of Cardiovascular Diseases and Internal Medicine, Mayo Clinic, Rochester, Minnesota, USA.
We report a case of restrictive cardiomyopathy in which a distinct endothelial thickening of the atrial wall and pulmonary vein orifices was noted on transesophageal echocardiography. Echocardiographically guided endomyocardial biopsy of the thickening revealed an inflammatory infiltrate that was rich in giant cells and provided important clues about an underlying immune mechanism for the pathogenesis. Positive results from the antineutrophil cytoplasmic autoantibody assay supported the diagnosis of Wegener's granulomatosis. After immunosuppressive therapy, the endomyocardial thickening completely resolved, but the restrictive cardiomyopathy did not reverse.
Ter Arkh. 1992;64(3):97-105.
Abramov's "primary myocarditis", Fiedler's myocarditis and dilated cardiomyopathy.
Novikov IuI, Stulova MA, Prokhorova IA.
After comparing the clinical and post mortem signs of myocarditis and dilated cardiomyopathy on the basis of correlating the reported data and own data on 28 patients with the appropriate descriptions by S. S. Abramov and A. Fiedler, the authors came to the conclusion that they had described two different diseases. S. S. Abramov had described dilated cardiomyopathy, whereas Fiedler acute diffuse myocarditis. The term Abramov-Fiedler myocarditis used in the Soviet literature does not correspond to the up-to-date classification of myocardial diseases. That is why it is required that these two concepts be separated to the benefit of the treatment.
Nervenarzt. 2005 May 20;
Diplopia and cardiogenic shock A case of orbital myositis associated with giant cell myocarditis.]
Kollmeier M, Brodhun M, Sliwka U, Sigusch H, Witte OW, Heide G.
Klinik fur Psychiatrie, Landeskrankenhaus Hildesheim, .
Idiopathic giant cell myocarditis is a rare and frequently fatal inflammatory heart disease which leads to congestive heart failure or ventricular arrhythmias. It is often associated with other autoimmune disorders. We report a 39-year-old woman who first presented with diplopia and painful eye movements, the typical clinical picture of orbital myositis. Shortly afterwards, she developed rapidly progressive congestive heart failure due to giant cell myocarditis, which took a fatal course within some weeks. Autopsy confirmed both disorders. This case report underlines the importance of early and repeated monitoring of cardiac function, if orbital myositis is suspected, in order to consider cardiac transplantation, the only efficacious treatment of giant cell myocarditis, in time.
Lupus. 2005;14(2):166-9.
Giant cell myocarditis: a rare cardiovascular manifestation in a patient with systemic lupus erythematosus.
Chung L, Berry GJ, Chakravarty EF.
Division of Immunology and Rheumatology, Department of Medicine, Stanford University School of Medicine, Palo Alto, CA 94304, USA.
Giant cell myocarditis (GCM) is a rare form of myocarditis with a median survival of less than one year. It has been reported to occur in patients with various underlying autoimmune diseases; however, no cases of GCM have been described in patients with clear evidence of underlying systemic lupus erythematosus (SLE). The presentation of GCM may mimic that of lupus myocarditis, including an initial response to immunosuppression. Despite initial clinical similarities, lupus myocarditis and GCM are histologically distinct entities with dramatic differences in prognosis. We report herein a patient with a longstanding history of SLE, who presented acutely with myocarditis, responded well to initial immunosuppression and then subsequently died of progressive heart failure that was found to be due to GCM. Endomyocardial biopsy can help define diagnosis and prognosis of lupus patients presenting with myocarditis, and early referral for cardiac transplantation should be considered in patients diagnosed with GCM.
Can J Cardiol. 2004 Apr;20(5):557-61.
Novel associations of giant cell myocarditis: two case reports and a review of the literature.
Shariff S, Straatman L, Allard M, Ignaszewski A.
Division of Cardiology, Saint Paul's Hospital and The University of British Columbia, Vancouver, Canada.
Giant cell myocarditis (GCM) is a rare and frequently fatal disorder with no proven treatment. It is a disease of young, predominantly healthy adults. Without transplantation, patients usually die of heart failure and ventricular arrhythmias. Due to the poor prognosis of the condition, prompt recognition and diagnosis are of clinical importance. Approximately 19% of these cases are associated with autoimmune diseases. The present article describes two unique cases of GCM associated with autoimmune diseases not previously reported in the literature - discoid lupus erythematosis and autoimmune hepatitis. A review of the natural history and treatment of GCM is also presented.
J Heart Lung Transplant. 2004 Apr;23(4):492-5.
Total lymphoid irradiation: new therapeutic option for refractory giant cell myocarditis.
Singh TP, Rabah R, Cooper LT, Walters HL.
Department of Pediatrics, Children's Hospital of Michigan, Wayne State University School of Medicine, Detroit, Michigan, USA. tsingh@dmc.org
Idiopathic giant cell myocarditis (GCM) is believed to be a T-lymphocyte-mediated autoimmune disease. Some patients with GCM have a dramatic clinical response to anti-T-cell immunosuppression. However, this response is not uniform and patients often deteriorate rapidly and need a cardiac transplantation within months of diagnosis. Following cardiac transplantation, GCM may recur in the graft but is usually mild and responds to augmentation of immunosuppression. This report is the first description of total lymphoid irradiation (TLI) for the treatment of GCM, which was used in a patient who developed an exceptionally early and severe recurrence of GCM in the cardiac graft that remained refractory to heightened immunosuppression for 4 months. Clinical and histologic remission followed a course of TLI and was maintained for 1 year despite a gradual decrease in immunosuppression. This novel treatment should be considered in all patients with GCM who do not have histologic remission with the currently employed anti-T-cell immunosuppression.
Angiology. 2002 Sep-Oct;53(5):599-603.
Coronary artery disease obscuring giant cell myocarditis--a case report.
Kwok OH, Chau EM, Wang EP, Chow WH.
Kwok Tak Seng Heart Centre, University Department of Medicine, Grantham Hospital, Aberdeen, Hong Kong. vohkwok@netvigator.com
A case in which the diagnosis of idiopathic giant cell myocarditis was obscured by the presence of severe coronary artery disease is described. A 47-year-old man presented with recurrent inferior myocardial infarction and complete heart block. Cardiac catheterization confirmed severe 2-vessel disease and left ventricular dysfunction. Incessant ventricular arrhythmia rapidly ensued, which did not respond to anti-arrhythmic therapy and overdrive pacing despite complete s urgical revascularization. He eventually died. Autopsy revealed giant cell myocarditis superimposed on coronary artery disease. Acute myocarditis masquerading as myocardial infarction has been well known, but virtually all reported cases had normal coronary arteries. This case illustrated the fact that even in the presence of obvious coronary artery disease the remote possibility of myocarditis should not be entirely disregarded. Although giant cell myocarditis is a rare and frequently fatal disorder, recent studies suggest that combined immunosuppressive therapy may improve the prognosis.
Herz. 2000 May;25(3):291-8.
Giant cell myocarditis: diagnosis and treatment.
Cooper LT Jr.
Division of Cardiovascular Diseases, Mayo Clinic, Rochester, MN, USA. cooper.leslie@mayo.edu
Giant cell myocarditis is a rare but devastating disease that usually affects young otherwise healthy individuals. Associations with thymoma, inflammatory bowel disease, and a variety of autoimmune disorders have been reported. The rate of death or heart transplantation is approximately 70% at 1 year. Data from a Lewis rat model and from observational human studies suggest that giant cell myocarditis is mediated by T lymphocytes and may respond to treatment aimed at attenuating T cell function. Recent findings from the Giant Cell Myocarditis Registry, a clinical and pathologic database from 63 cases of giant cell myocarditis gathered from 36 medical centers, include the following: The sensitivity of endomyocardial biopsy for giant cell myocarditis for patients who undergo transplantation or autopsy is 82 to 85%. Registry subjects who received cyclosporine in combination with steroid, azathioprine, or muromonab-CD3 have prolonged transplant-free survival (12.6 months vs. 3.0 months for no immunosuppression). Post-transplantation survival is approximately 71% at 5 years despite a 25% rate of giant cell infiltration in the donor heart. To confirm and extend these findings, a randomized trial of immunosuppression including muromonab-CD3, cyclosporine, and steroids is underway.
J Heart Lung Transplant. 2001 Mar;20(3):375-80.
Recurrence of giant cell myocarditis in cardiac allograft.
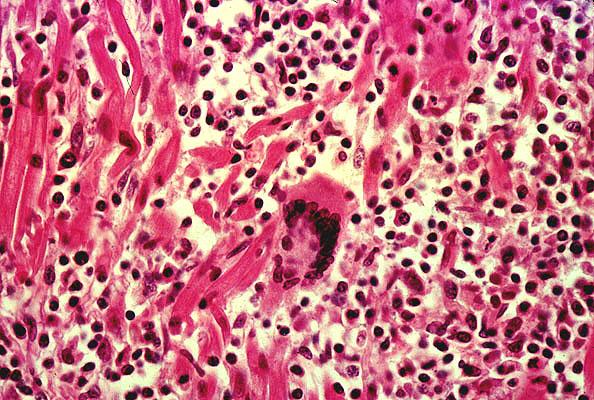
Scott RL, Ratliff NB, Starling RC, Young JB.
Ochsner Heart and Vascular Institute, New Orleans, Louisiana 70121, USA. rscott@ochsner.org
BACKGROUND: Giant cell myocarditis causes essentially irreversible fulminant left ventricular dysfunction with associated conduction abnormalities and congestive failure. Response to immunosuppressive therapy is poor and cardiac transplantation is the only viable treatment option. The histologic hallmarks of giant cell myocarditis include a polymorphous inflammatory response with numerous multinucleated giant cells and extensive myocyte necrosis in a geographic pattern. There were 38 patients who received a cardiac transplant for giant cell myocarditis in the Giant Cell Myocarditis Registry. Among these patients, there were 9 recurrences of disease in the allograft. Concern has been expressed that recurrence of giant cell myocarditis in the allograft might be a contraindication for cardiac transplantation in the future. METHODS: In our single-center analysis we describe the clinical and histologic findings of 5 patients transplanted for giant cell myocarditis at the Cleveland Clinic. RESULTS: All but 1 of the patients were New York Heart Association (NYHA) class 4 with an average cardiac index (CI) of 1.52 liters/min x m(2). Of the 5 patients transplanted, 1 developed recurrent giant cell myocarditis. Routine right ventricular endomyocardial biopsy at 1 week exhibited severe multifocal myocardial fibrosis in addition to mild acute vascular rejection and mild grade 1A cellular rejection. Follow-up biopsy in this patient indicated grade IIIA moderate acute rejection in addition to multinucleated giant cells. Two distinct inflammatory processes were noted consisting of foci of T-cell inflammation identified by immunohistochemistry to be consistent with rejection, and a second inflammatory process with few mononuclear cells staining for macrophage or T-cell markers with eosinophils and myocyte necrosis consistent with giant cell myocarditis. Follow-up right ventricular endomyocardial biopsies (RVBXs) in this patient have subsequently demonstrated improvement in the degree of inflammatory infiltrate without vascular or significant cellular rejection. Vascular rejection was noted in 1 of the remaining 4 patients and was treated successfully with muramab-CD3 and plasmapheresis. CONCLUSIONS: Giant cell myocarditis should be expected to recur in the allograft and often does so concurrently with rejection. However, the disease in the allograft responds to therapy in a favorable manner, which differs dramatically from that in the native heart. This might be the result of detection of the disease at an earlier stage than in the native heart, or the immunosuppression milieu in the allograft. The favorable response to therapy suggests that the likelihood of recurrence of giant cell myocarditis should not be considered a barrier to transplantation.
Survival Outcomes of Patients with Giant Cell Myocarditis Bridged by Ventricular Assist Devices.
ASAIO Journal. 46(5):569-572, September/October 2000.
Brilakis, Emmanouil S.; Olson, Lyle J.; Berry, Gerald J.; Daly, Richard C.; Loisance, Daniel; Zucker, Mark; Cooper, Leslie T. Jr.
ABSTRACT:
Giant cell myocarditis is a highly lethal disorder characterized by rapidly progressive congestive heart failure. The aim of this study was to describe the clinical course of patients with giant cell myocarditis who received a ventricular assist device. Patients with giant cell myocarditis were identified from the Multicenter Giant cell Myocarditis Registry. Bridging to cardiac transplantation in the giant cell myocarditis patients who received a ventricular assist device was compared with bridging in the general population of heart failure patients, as reported in the literature. Median posttransplantation survival for patients with giant cell myocarditis who received and did not receive ventricular assist devices was calculated by the Kaplan-Meier method and compared with use of the log-rank test. Nine patients with giant cell myocarditis who received ventricular assist devices were identified. Seven patients survived to transplantation, four were alive 30 days posttransplantation, and two survived to 1 year. The rate of successful bridging to transplantation in seven of nine patients (78%) is similar to that reported for other ventricular assist device recipients. Posttransplantation survival of 57% (4 of 7) at 30 days and 29% (2 of 7) at 1 year was significantly lower compared with 93% 1-year survival of the 30 patients with giant cell myocarditis who did not receive ventricular assist devices before transplantation (p < 0.001). Ventricular assist devices can be an effective bridge to transplantation for patients with heart failure caused by giant cell myocarditis. Although their posttransplantation survival was poor in our series, a few patients had long-term survival.
Eur J Cardiol. 1978 Oct;8(3):349-58.
Complete heart block due to granulomatous giant cell myocarditis: report of 3 cases.
Lindvall K, Edhag O, Erhardt LR, Sjogren A, Swahn A.
3 patients with chronic complete AV block were found at autopsy to have granulomatous giant cell myocarditis (GGCM). In 1 patient an unusual clinical course led to more extensive investigation including echocardiography which revealed ventricular septal abnormalities. A review of the literature is presented. Although GGCM is a rare disease echocardiography may be a useful screening procedure in patient with AV block especially in the presence of immunological disorders.
Kokyu To Junkan. 1991 Apr;39(4):373-6.
A case of giant cell myocarditis associated with a progressive disturbance in the conduction system
Saijo Y, Nitta S, Katahira Y, Takahashi K, Yambe T, Endoh N, Sonobe T, Tanaka M, Meguro T, Nitta K.
Department of Medical Engineering and Cardiology, Tohoku University.
A 73 year old male patient with a history of pulmonary tuberculosis was admitted to our department because of dyspnea and abdominal pain. The chest X-ray film on admission showed bilateral lung congestion. The ECG showed atrial fibrillation, left axis deviation and incomplete right bundle branch block. Five days after admission, the ECG changed into sinus rhythm and complete right bundle branch block. Eight days after admission, the patient complained of chest pain and the ECG showed ST elevation in II, III, aVF, reciprocal ST depression in V, and complete A-V block with junctional rhythm. Emergency coronary angiography revealed no significant stenosis. Echocardiography showed reduced contraction of the inferior wall and diffuse granular echoes in the myocardium. Light microscopic study revealed fibrosis, infiltration of eosinophils and histiocytes, degenerated myocardium and multinucleated giant cells. Some of the giant cells were morphologically similar to myocardium, so the myocardium might be a place of immunological reaction.
Support with the BVS 5000� Assist Device during Treatment of Acute Giant-Cell Myocarditis
Daniel Marelli, MD, Reza Kermani, BS, Jessica Bresson, BS, Michael C. Fishbein, MD, Michele Hamilton, MD, Jaime Moriguchi, MD,
Gregg C. Fonarow, MD, Benjamin Cohen, MD, Jon Kobashigawa, MD, Hillel Laks, MD (Los Angeles)
Giant-cell myocarditis is a rare and aggressive form of myocarditis with a high mortality rate. Our purpose is to summarize 3 cases of acute giant-cell myocarditis that illustrate possible outcomes with mechanical support.
We reviewed the cases of 3 patients, aged 39 to 59 years, who had giant-cell myocarditis (confirmed by myocardial biopsy). The indication for ventricular assist was circulatory failure despite maximal medical treatment with 2 or more inotropic agents and intraaortic balloon pump support. Immunosuppression and a biventricular mechanical assist (BVS 5000) were used to treat all these patients. The mean duration of mechanical support was 15.7 days (range, 10 to 19 days).
One patient had recovery of myocardial function and was weaned from mechanical support. This case is, to our knowledge, the first reported of ventricular support leading to cardiac recovery after diagnosis of giant-cell myocarditis. The 2nd patient was not a candidate for further surgery and died of multisystem organ failure. The 3rd patient underwent orthotopic heart transplantation after 18 days of support and was discharged.
We conclude that patients with giant-cell myocarditis tend to have biventricular involvement and can recover myocardial function on mechanical support and immunosuppression. If recovery is not observed, transplantation is warranted. By avoiding left ventricular cannulation, the BVS 5000 is well suited for bridging to recovery, transplantation, or long-term support. (Tex Heart Inst J 2003;30:50-6)
Chest. 2000 Mar;117(3):905-7.
Giant cell myocarditis responding to immunosuppressive therapy.
Frustaci A, Chimenti C, Pieroni M, Gentiloni N.
Departments of Cardiology, Catholic University, Rome, Italy.
An unusual case of giant cell myocarditis presenting with cardiogenic shock that dramatically responded to conventional dose of steroids and azathioprine is reported. Cardiac recovery was rapid, complete (left ventricular ejection fraction rose to 55% from 10%), and was accompanied by the disappearance of the inflammatory infiltrates including giant cells in the control endomyocardial biopsy. Maintenance of the recovery at 16 months of follow-up on a low dose of azathioprine suggests that giant cell myocarditis might be a heterogeneous disease having either a negative untreatable trend necessitating cardiac transplantation, or a curable substrate responding to immunosuppressive drugs.
N Engl J Med. 1997 Jun 26;336(26):1860-6.
Idiopathic giant-cell myocarditis--natural history and treatment. Multicenter Giant Cell Myocarditis Study Group Investigators.
Cooper LT Jr, Berry GJ, Shabetai R.
Department of Medicine, University of California at San Diego Medical Center, USA. cooper.leslie@mayo.edu
BACKGROUND: Idiopathic giant-cell myocarditis is a rare and frequently fatal disorder. We used a multicenter data base to define the natural history of giant-cell myocarditis and the effect of treatment. METHODS: We identified 63 patients with idiopathic giant-cell myocarditis through journal announcements and direct mailings to cardiovascular centers worldwide. RESULTS: The patients consisted of 33 men and 30 women with an average age of 42.6 years; 88 percent were white, 5 percent were black, 5 percent were Southeast Asian or Indian, and 2 percent were Middle Eastern. Most presented with congestive heart failure (47 patients, or 75 percent), ventricular arrhythmia (9 patients, or 14 percent), or heart block (3 patients, or 5 percent), although in some cases the initial symptoms resembled those of acute myocardial infarction (4 patients). Nineteen percent had associated autoimmune disorders. The rate of survival was worse than among 111 patients with lymphocytic myocarditis in the Myocarditis Treatment Trial (P<0.001); among our patients, the rate of death or cardiac transplantation was 89 percent, and median survival was only 5.5 months from the onset of symptoms. The 22 patients treated with corticosteroids and cyclosporine, azathioprine, or both therapies survived for an average of 12.3 months, as compared with an average of 3.0 months for the 30 patients who received no immunosuppressive therapy (P=0.001). Of the 34 patients who underwent heart transplantation, 9 (26 percent) had a giant-cell infiltrate in the transplanted heart and 1 died of recurrent giant-cell myocarditis. CONCLUSIONS: Giant-cell myocarditis is a disease of relatively young, predominantly healthy adults. Patients usually die of heart failure and ventricular arrhythmia unless cardiac transplantation is performed. Despite the possibility of fatal disease recurrence, transplantation is the treatment of choice for most patients.
Back to E-chocardiography Home Page.
e-mail:shindler@umdnj.edu
The contents and links on this page were last verified on May 27, 2005.
{kind=link}